
Advanced Computing, Mathematics and Data
Research Highlights
March 2008
New Software Developed for Remote Protein Homologs Is Significantly More Sensitive than Current Tools

Anuj Shah
{kind=link}
Researchers at Pacific Northwest National Laboratory have developed an innovative software tool to detect homologs (proteins with similar function but dissimilar sequence) that significantly outperforms most current methods for remote homolog identification.
"As the amount of biological sequence data continues to grow exponentially, we face the increasing challenge of assigning function to this enormous molecular ‘parts list,' said Anuj Shah, the lead developer for SVM-HUSTLE. "The most popular approaches to meet this challenge make use of the simplifying assumption that similar functional molecules, or proteins, sometimes have similar composition, or sequence. However, these algorithms often fail to identify remote homologs (homologs that have low sequence identity) that often are a significant fraction of the total homolog collection for a given sequence."
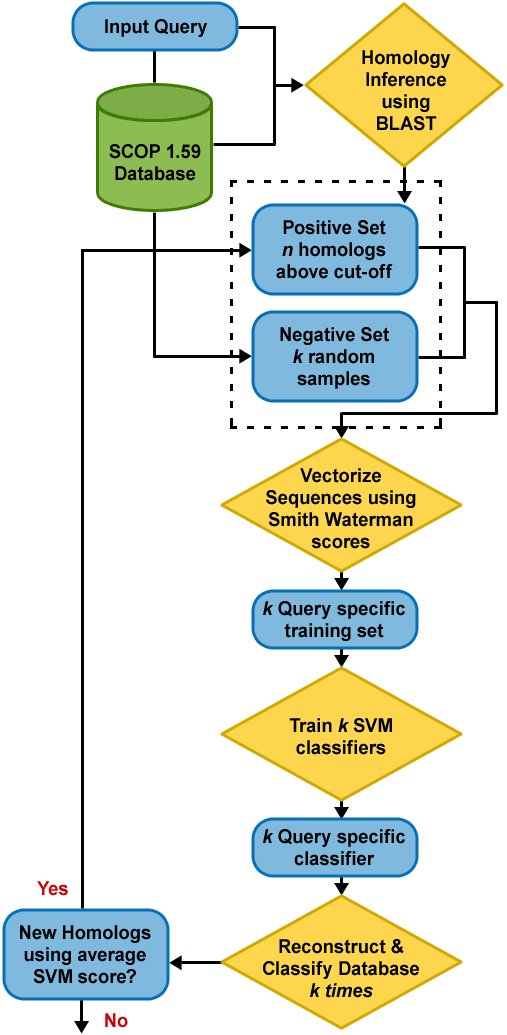
Shah, along with PNNL's Christopher Oehmen and Bobbie-Jo Webb-Robertson, introduced SVM-HUSTLE (Support Vector Machine-based tool to detect Homology Using Semi-supervised iTerative LEarning) that identifies significantly more remote homologs than current state-of-the-art sequence or cluster-based methods. Their research paper was recently published in Bioinformatics.
As opposed to building profiles or position specific scoring matrices, SVM-HUSTLE builds an SVM classifier for a query sequence by training on a collection of representative high-confidence training sets, recruits additional sequences and assigns a statistical measure of homology between a pair of sequences. SVM-HUSTLE combines principles of semi-supervised learning theory with statistical sampling to create many concurrent classifiers to iteratively detect and refine, on-the-fly, patterns indicating homology.
When compared against existing methods for identifying protein homologs (BLAST, PSI-BLAST, COMPASS, PROF_SIM, RANKPROP and their variants) on two different benchmark datasets SVM-HUSTLE significantly outperforms each of the above methods. SVM-HUSTLE also yields results comparable to HHSearch, a method that uses profile-profile comparison, but at a substantially reduced computational cost.
The software executable to run SVM-HUSTLE can be downloaded.
Acknowledgment
Sponsors—The research was supported by the Data Intensive Computing Initiative Laboratory Directed Research and Development Program at the Pacific Northwest National Laboratory, (PNNL) and under the Data-intensive Computing for Complex Biological Systems project funded by the Office of Advanced Scientific Computing Research. PNNL is a multi-program national laboratory operated by Battelle for the U.S. DOE under contract DE-AC06-76RL01830.
EMSL Involvement—The research was performed in part using the Molecular Science Computing Facility (MSCF) in the William R. Wiley Environmental Molecular Science Laboratory (EMSL) under EUS computational grand challenge project 20905 and the PNNL Advanced Computing Center (PACC) under EUS project pacc63. EMSL is a national scientific user facility sponsored by the U.S. DOE, OBER and located at PNNL.
Reference
SVM-HUSTLE - An iterative semi-supervised machine learning approach for pairwise protein remote homology detection. Shah AR, Oehmen CS, Webb-Robertson BJ. Bioinformatics. 2008 Mar 15;24(6):783-90. Epub 2008 Feb 1. PMID: 18245127 [PubMed - in process]