
Advanced Computing, Mathematics and Data
Research Highlights
February 2014
Better Chemistry Through Computing
Parallel in time algorithms speed up molecular simulation time scales
Results: As computers continue to grow into massively parallel super systems, it is becoming possible to address the chemical complexity and time scale of problems likely to be encountered in new energy strategies, such as nuclear waste storage, carbon sequestration, energy storage materials, and efficient catalysis, using simulations that include fundamental atomic and molecular interactions. Unfortunately, most chemical processes occur on time scales out of reach of current molecular simulations as these evolve by integrating over time in serial, which results in simulations that are not long enough to capture realistic chemical phenomena. To conduct longer molecular simulations in areas such as aerosol chemistry and human health more efficiently, scientists from the Environmental Molecular Sciences Laboratory (EMSL) at Pacific Northwest National Laboratory (PNNL); the University of Chicago; and the University of California, San Diego, developed and assessed parallel in time algorithms, where forward integration of Newton equations is distributed over many processors and applied to atomic-level molecular dynamics (MD) or ab initio molecular dynamics (AIMD) simulations.
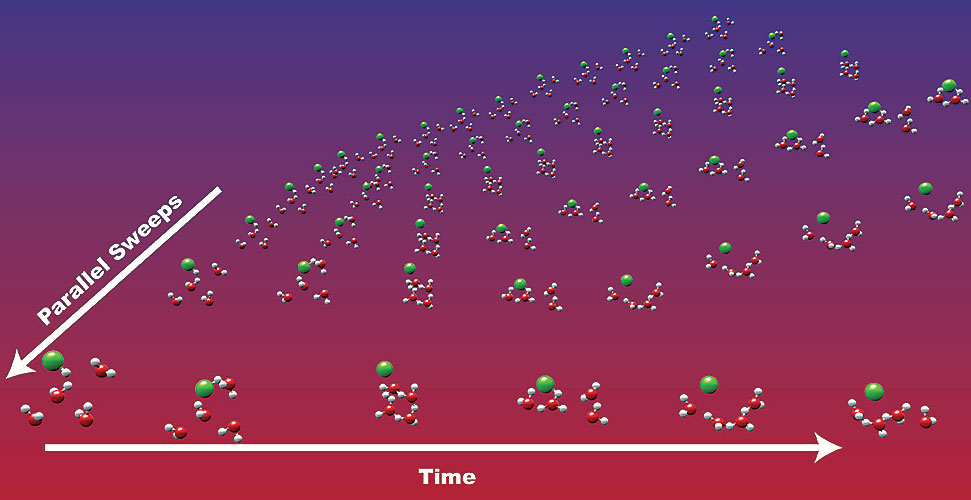
New parallel in time algorithms speed up high-level molecular dynamics simulations to enable predictions involving the properties of complex materials. Enlarge Image.
Why it Matters: Parallel in time algorithms can expand the time scale of calculations once limited by scaling. With these timings, meaningful dynamic simulations of realistic, complex materials phenomena or problems can be demonstrated using intricate computer simulations of complex physical motions of atoms and molecules. Moreover, these timings afford meaningful-and real-world applicable-dynamic simulations in research areas as diverse as environmental remediation, carbon sequestration, energy production and storage, human health issues, catalysis, and other applications relevant to the nation's major energy and environmental problems.
Methods: With similar size and complexity to actual materials chemistry problems, scientists tested a 1000 silicon atom liquid MD simulation using Stillinger-Weber potentials (represents crystal lattices) and an HCl+4H2O AIMD simulation at the Møller-Plesset second-order perturbation (MP2) level of theory. For the parallelized Stillinger-Weber MD simulation, the algorithm achieved a maximum speedup of 3.0, while the MP2 level used in the AIMD demonstrated speedups up to 14.3. In both cases, the speedups occurred across slow networks, indicating these algorithms can be used for long time dynamics AIMD simulations at a modest cost with machines connected via WiFi or in different time zones by the Internet. These algorithms offer reasonable speedup even when accurate and inexpensive coarse modes are not available and can be easily implemented in scripting languages (e.g., Python) that call to standard quantum chemistry packages, such as NWChem.
What's Next? The authors acknowledge that for some problems, parallel in time approaches may need to be recast from a simple master-slave parallel implementation to a complex dataflow parallel implementation. While their work obtains modest speedups without complex preconditioners, the development of more complex preconditioners based on extended spatial scales or softer potentials (likely a difficult task) probably will be needed to reach realistic time scales. They also consider another approach that involves using algorithms based on implicit integrators, which may resolve the multiple time scales in a hierarchal fashion (as with multigrid solvers) and overcome broadcast bottlenecks.
Acknowledgments: This research was supported by the U.S. Department of Energy (DOE), Office of Advanced Scientific Computing Research Petascale Tools program and DOE's Office of Basic Energy Sciences Geosciences program, as well as through an award from the National Science Foundation. DOE's Office of Energy Research provided computer time at the National Energy Research Scientific Computing Center. Some of the calculations also were performed using EMSL (Chinook and Spokane computing systems), a national scientific user facility sponsored by DOE's Office of Biological and Environmental Research and located at PNNL.
Reference: Bylaska EJ, JQ Weare, and JH Weare. 2013. "Extending molecular simulation time scales: Parallel in time integrations for high-level quantum chemistry and complex force representations." Journal of Chemical Physics 139(7):074114. DOI: 10.1063/1.4818328.
Related Highlight: Just in Time